通过机器学习和混合特征选择方法预测蛋白质中赖氨酸丙二酰化位点
题目:a hybrid feature selection method for predicting lysine malonylation sites in proteins via machine learning
期刊名称:Cemometrics and Intelligent Laboratory Systems
影响因子:4.175
发表时间:
摘要
本文将几种特征选择方法相结合,提出了一种新的特征选择方法。首先给出了常用的特征提取方法EAAC、EGAAC、PKa、TF-IDF、TF-CRF和PSSM的算法,然后给出了这些算法的组合模型。提出的方法已经在小鼠、人类和大肠杆菌的三个赖氨酸丙二酰化数据集上进行了预测,同时还使用了几种机器学习方法对数据进行分类。最后,为了证明该方法的有效性,作者计算了一些重要的参数,并与其他特征提取方法进行了比较。
简介
准确识别丙二酰化位点非常重要,可以为生物医学研究和更好地理解分子功能提供有用的消息。然而实验检测丙二酰化位点主要是通过质谱法完成的,比较耗时耗力。计算方法可以用来更有效和准确地确定丙二酰化位点。本研究的主要目的是介绍从蛋白质中提取特征的几种组合方法,以识别丙二酰化位点。
实验说明
1、序列分析
根据蛋白质的序列分析,大肠杆菌、智人和小鼠数据集的丙二酰化和非丙二酰化位点的审定差异见图1。
图1. 氨基酸在富集区和贫化区的分布及其与中心赖氨酸的比较
它显示了来自样本蛋白质的氨基酸的分布,并描述了位于赖氨酸周围的氨基酸的其他剩余部分的范围(+12)至(–12)。在不同的赖氨酸分子中,丙二酰化和非丙二酰化位点的氨基酸自由度有显著的差异。与中央赖氨酸相比,选择性氨基酸分为富集和贫化两部分。据此可以窥察到,在富集段赖氨酸周围氨基酸的剩余部分频率高于其他部分,且离中心赖氨酸越远,富集段频率比越低。此外,图1强调了基于序列蛋白的特征提取的重要性,因为靠近中央赖氨酸的氨基酸的其余部分的位置分离得很好。因此,我们需要不同的特征提取方法来更好地识别丙酰化部位。
2、提出的方法
本文提出了一个预测丙二酰化位点的新模子,如图2所示。
图2. 建议的丙酰化和非丙酰化部位分类方法的步骤
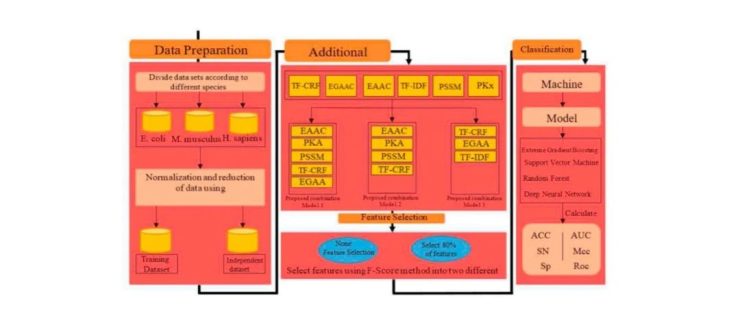
实验步骤
由六个主要步骤组成,包括数据集的选择、特征提取、特征归一化、特征组合、特征选择,最后是模型性能评估。(1)选择数据集。三个数据集大肠杆菌、智人和小鼠被用来学习和测试。使用的数据集被随机分为训练数据集和测试数据集。可在https://github.com/QUSTAIBBDRC/DeepMal/tree/master/Datasets中获得。(2)特征提取。本文中使用的特征提取方法包括EAAC、EGAAC、TFIDF、PSSM、PKa和TFCRF。(3)特征归一化。为了提高模型的性能,必须对数据进行标准化处理。Z-Score方法被用来对特征进行规范化处理。(4)结合特征。在审查不同的数据和特征时,考虑了不同特征的组合可能会提高分类器模型的检测率的想法。为了研究这个问题,这些特征以不同的模式进行了组合。在第一个模型中,(TFCRF、EGAAC、TFIDF)的特征,产生的特征向量长度包括39个特征;在第二个模型中,(EAAC、PKA、PSSM、TFCRF)的特征,得到的特征向量长度包括487个特征;在第三个模型中,(EAAC、PKA、PSSM、TFCRF、EGAAC)的特征被组合,得到的特征向量长度包括590个特征。(5)特征选择。采用了FScore方法,如果一个特征的Fscore值很高,表明该特征具有良好的分类信息。(6)模型性能评估。本研究使用十折交叉验证法来评估分类模型的预测性能。本研究中使用的各种分类方法包括梯度提升(XGBoost)、支持向量机(SVM)、随机森林(RF)、深度神经网络(DNN)。计算了AUC、ACC、Sn、Sp和MCC的值。此外,为了更准确地评估,还绘制了ROC曲线和条形图以及误差和其他分析图表。
数值结果
使用这三个模型中的组合特征进行不同类别的训练。表1显示了这个实验在不同分类模型上的结果,XGboost算法在所有建议的组合模型中表现更好,并且比其他算法有更高的准确性。
表1 三种模型对三个数据集预计丙二酰化位点的性能比较
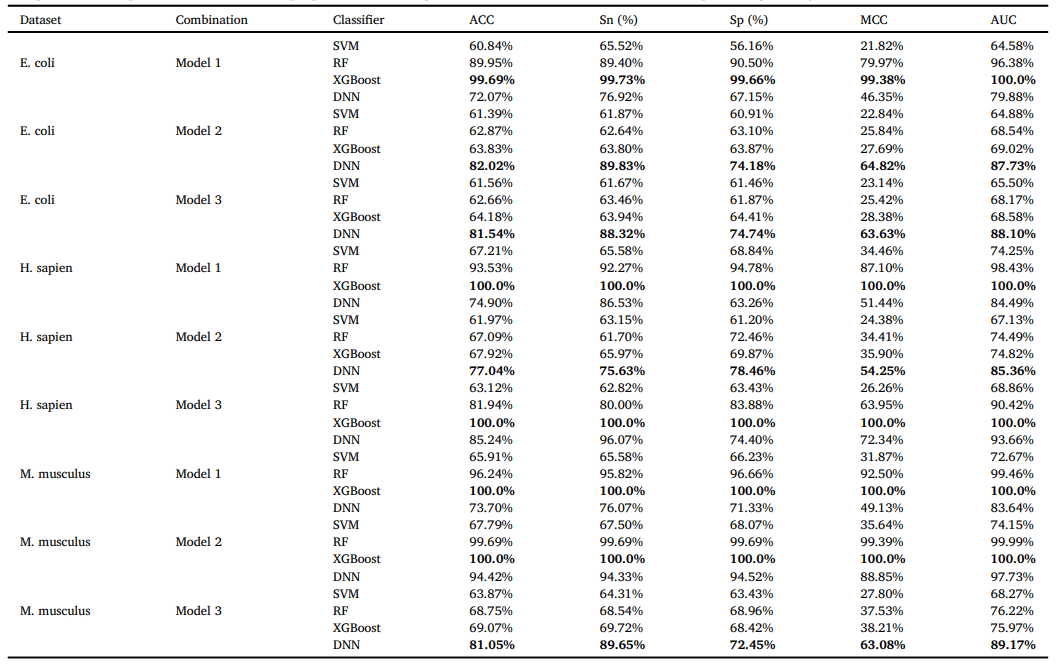
表2 比较三种提出的模式的性能,使用F-Score方法选择一个特征来预测三个数据集的丙二酰化位点
表2列出了数据集和不同的分类方法。在fscore法子上得分最高的80%的特征被选中。
表格显示,所提出的模型中提取和组合的所有特征的高度重要性。
为了更好地分析提议的模型,图3表现了不同类别上的roc曲线。
图3. 在第一个提出的模型中,具有80%的最佳特征,不同分类算法的预测性能
在该图中,ROC曲线显示在第一个提出的模型中,其中80%的最佳特征来自智人、大肠杆菌和小鼠的3个学习数据集。从SVM、XGBoost、RF和DNN分类器的ROC曲线可以看出,XGBoost分类器的ROC曲线下的面积明显高于其他三种分类算法,说明XGBoost的算法分类对赖氨酸蛋白丙二酰化和非丙二酰化位点具有很强的概括能力和良好的预测性能。
为了评估各方法提取的特征,基于k10的交叉验证,使用eaac、egaac、pka、tfidf、tfcrf和pssm的各特色训练默认rf模型。这个实验的结果显示在图4中的三个数据集上。
图4. 对三个数据集使用6种差别特征类型的训练rf模型的性能进行比较
从图4中可以看出,通过TFCRF提取的特征在三个数据集上都有较高的检测率。此外,EAAC、EGAAC和PKa方法的性能也可以接受。根据每个数据集的第一、第二和第三模型的拟议组合结果,很明显,不同特征的组合可以提高分类器模型的检测率。
为了显示模型的强度和稳定性,我们使用误差分析。误差条表示估计的缺点或不确定性,以获得对测量精度的总体了解(图5)。
图5. 使用误差条对大肠杆菌、智人和小鼠分类模子的研究
在图5中,xgboost、rf、svc和dnn等算法挨次表达了较高的精度结果。
与其他方法的比较:为了进一步分析所提出的方法,与其他不同的方法进行了比较,在大肠杆菌、智人和小鼠的数据集中进行了预测,并绘制了评估指标ACC、SN、SP和MCC量的柱状图。如图6所示,
图6. 提出的方法与其他法子在三个数据集上的比较
本文链接: https://www.yizhekk.com/0119256457.html







