jcim|激增的机器学习法子推动qsar研究的再发展

11月28日,journal of chemical information and modeling期刊编辑部发表了一篇评论性论文the (re)-evolution of quantitative structure–activity relationship (qsar) studies propelled by the surge of machine learning methods。
论文中,作者回顾了jcim期刊上发表的qsar的机器学习方法,讨论了qsar研究的进展,为研究者精选了一系列典范研究论文,希望能够吸引研究者为qsar研究再添新进展,进一步推动qsar与机器学习方法的关联研究。
1962年,在hansch及其同事所做的关于定量结构-活性关系(quantitative structure−activity relationship, qsar)的开创性职业中,预测hammett函数和分配系数将在建立构效关系中变得非常重要。在过去的60年中,qsar已经从小数据集的类似化合物的粗糙回归/分类分析发展到基于精巧的机器进修技术,机器学习可以提取嵌入在复杂结构的分子组成的大数据集中的化学、物理和生物功能信息特征。通过结构-活性映射关系的转换,qsar成为药物发现的重要组成部分。这使得研究者可以高效、低成本地预测分子活性和性质,以及基于结构的虚拟筛选数百万候选药物组成的化学库得到有潜力的hits。机器学习也应用于各种其他领域,包括化合物的逆向合成路线预测,蛋白质和化合物设计,构象生成,力场优化和蛋白质结构预测。经典的qsar方法依赖于数学模型来建立各种描述符与生物活性之间的瓜葛。这些描述符包括分子指纹、图或其他数学表示等。生物活性包括吸收、分布、代谢、排泄、毒性(admet),结合自由能,蛋白质-配体复合物的动力学速率等。建立这样的关系也常常需要基于具有相似拓扑结构和功能的分子组成的数据集。由于可以使用广泛的数学模型,qsar很早就结合了机器学习算法,通过多任务模型建模非线性结构-功能关系来处理大且高维数据集。
值得注意的是,包括深度学习在内的机器学习最近的发展已经远远超出了QSAR。例如,基于高级自然语言处理(natural language processing, NLP)的自编码器提供来自未标记数据的序列嵌入,用于准确的分子性质预测。此外,transformer利用自监督学习(self-supervised learning, SSL)策略学习数亿分子和生物分子序列信息,并实现无需结构的分子活性预测。在任务方面,机器学习和深度学习不仅涉及回归和分类,还涉及聚类和降维,这在组学数据分析中有着广泛的应用。大量的机器学习和深度学习技术已经被开发并应用于化学科学,包括生成对抗网络(GAN)、U-Net、长短时记忆神经网络(LSTM)、图神经网络(GNN)、强化学习(RL)、玻尔兹曼机(Boltzmann machine)等。尽管迁移学习仍然是一个流行的问题,但主动学习已被应用于化学信息。各种机器学习和深度学习策略被开发出来,以在不利条件下提取化学特征,不利条件包括但不限于:多源数据、不平衡数据、噪声数据和小数据等条件。化学信息学和建模领域已经超越了化学领域,研究者来自计算机科学、数学、化学、生物和/或医学工程行业等等。
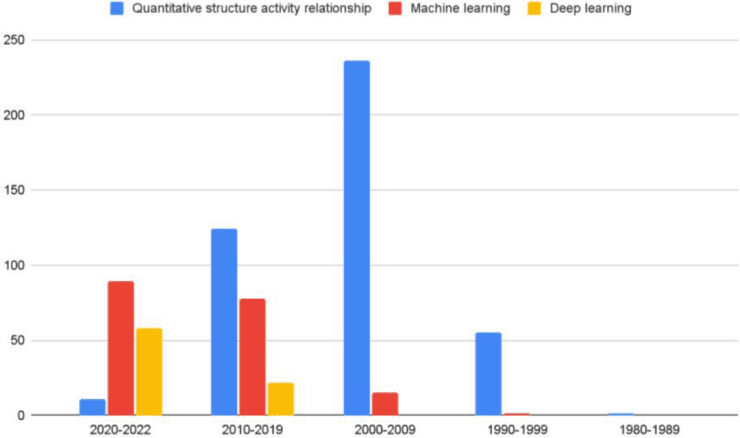
图1 “quantitative structure activity relationship”、“machine learning”和“deep learning被JCIM上出版的论文提及次数的分年份统计图
参考文献
Soares T A, Nunes-Alves A, Mazzolari A, et al. The (Re)-Evolution of Quantitative Structure–Activity Relationship (QSAR) Studies Propelled by the Surge of Machine Learning Methods[J]. Journal of Chemical Information and Modeling, , 62(22): 5317-5320.
本文链接: https://www.yizhekk.com/0147246668.html







